The intricate and often baffling world of blood disorders is intrinsically woven into the fabric of human genetics. Far from being random occurrences, a substantial number of hematological conditions—those affecting the blood, bone marrow, and lymphatic system—are direct consequences of variations, deletions, or single-point mutations within the human genome. These conditions are a powerful reminder that our biological blueprint, housed within the nucleus of every cell, dictates the efficiency and architecture of our entire circulatory system. The process of hematopoiesis, the continuous formation of blood cellular components, is a tightly regulated cascade, and even a minor error in the instructional code can lead to profound, systemic dysfunction. To fully grasp the nature of these diseases, one must move beyond the clinical symptoms—anemia, bleeding, or clotting—and delve into the molecular-level machinery that underlies the production, structure, and function of red cells, white cells, and platelets. This field of study, often perceived as highly technical, is where the foundational secrets of inherited illness reside, demonstrating how a single base-pair change can alter a patient’s life trajectory. The seemingly simple phrase “blood disorder” conceals a complex array of biological failures, each traceable back to a distinct genetic origin.
A substantial number of hematological conditions… are direct consequences of variations, deletions, or single-point mutations within the human genome.
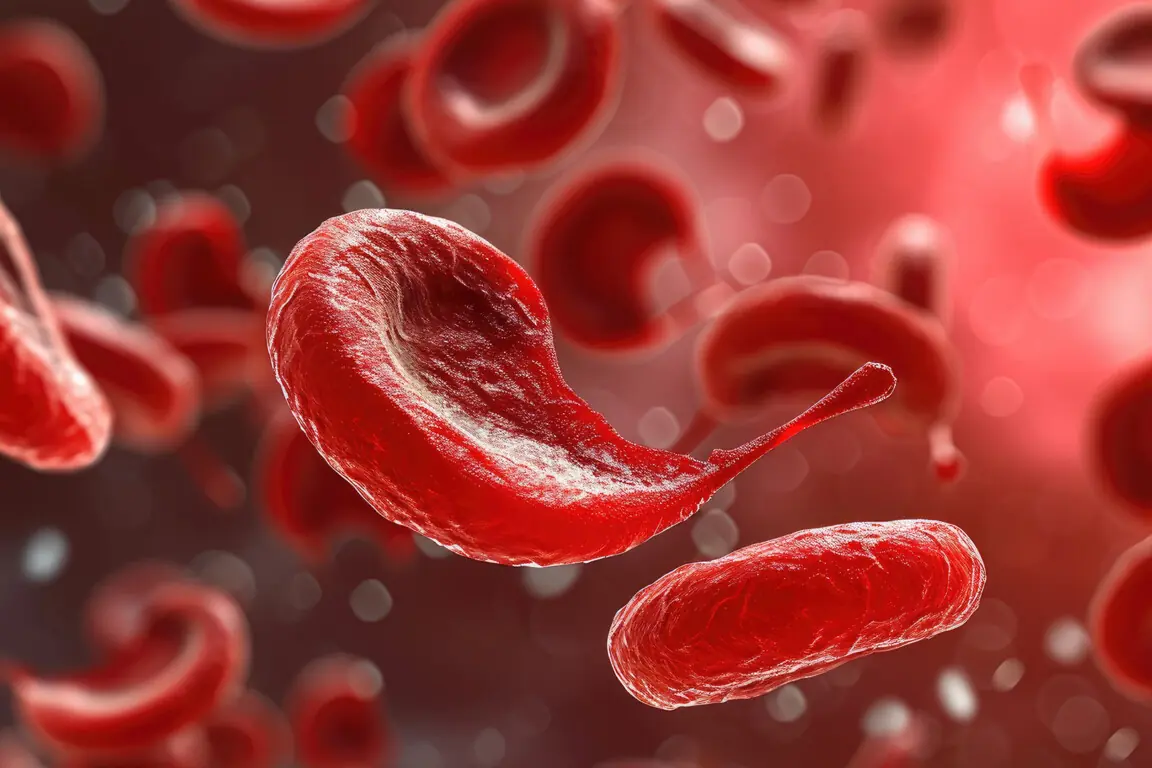
The primary types of inherited blood disorders generally fall into three broad categories: hemoglobinopathies, which affect the oxygen-carrying protein hemoglobin; coagulopathies, which disrupt the complex cascade of blood clotting; and inherited bone marrow failure syndromes, which impair the body’s ability to produce blood cells effectively. Hemoglobinopathies, such as Sickle Cell Disease (SCD) and the various thalassemias, are perhaps the most widely recognized globally. Sickle Cell Disease, for instance, results from a single base-pair substitution—adenine for thymine—in the -globin gene (
) on chromosome 11. This minuscule error replaces glutamic acid with valine in the
-chain of hemoglobin, fundamentally destabilizing the red blood cell structure when deoxygenated. The resulting sickle shape is not just a curiosity; it is the physical manifestation of a genetic coding error that leads to chronic hemolysis, vaso-occlusive crises, and organ damage. The geographic distribution of these disorders, historically concentrated in malaria-endemic regions, further illustrates a fascinating but grim evolutionary trade-off, where carrying a single copy of the faulty gene provided a selective advantage against a pervasive infectious disease.
Sickle Cell Disease, for instance, results from a single base-pair substitution—adenine for thymine—in the ![]() -globin gene (
-globin gene (![]() ) on chromosome 11.
) on chromosome 11.
Thalassemias represent another major group of hemoglobinopathies, characterized by a reduced rate of synthesis of one of the globin chains ( or
). Unlike the structural defect in SCD, thalassemia involves a quantitative defect in production.
-thalassemia major, a severe form, arises when an individual inherits defective
-globin genes from both parents, leading to a profound deficiency of mature red cells and chronic, life-threatening anemia that necessitates lifelong blood transfusions. The genetic causes are highly heterogeneous, involving dozens of possible point mutations, small deletions, or splice-site mutations in the
gene.
-thalassemia, conversely, is usually caused by the deletion of one or more of the four
-globin genes on chromosome 16. The severity of the condition correlates directly with the number of deleted genes, ranging from the asymptomatic carrier state (one deletion) to Hemoglobin Bart’s Hydrops Fetalis (four deletions), which is generally incompatible with life. This spectrum highlights how subtle differences in the type and location of a genetic alteration can dramatically alter the clinical presentation and prognosis, making classification and genetic counseling an extraordinarily complex task.
The genetic causes are highly heterogeneous, involving dozens of possible point mutations, small deletions, or splice-site mutations in the ![]() gene.
gene.
Moving away from red cell components, inherited coagulopathies present a very different, yet equally life-altering, set of challenges rooted in the genes responsible for the delicate balance of hemostasis. The most notable example is Hemophilia, an X-linked recessive disorder primarily affecting males. Hemophilia A, the more common form, involves a deficiency in functional Factor VIII, a critical protein in the intrinsic clotting pathway, while Hemophilia B is characterized by a Factor IX deficiency. Both are caused by mutations in the respective genes, and
, located on the X chromosome. The specific mutation types—ranging from large gene inversions to single-nucleotide variants—are highly diverse and directly influence the severity of the bleeding phenotype. The genetic mapping of these factors has not only enabled precise diagnosis and carrier testing but also paved the way for recombinant protein therapy, a medical triumph directly born from molecular genetic understanding. However, the genetic complexity does not end with these well-known examples; a multitude of rare, less-understood factor deficiencies (e.g., Factor XI deficiency, von Willebrand Disease) contribute to the mosaic of inherited bleeding tendencies.
The most notable example is Hemophilia, an X-linked recessive disorder primarily affecting males.
Inherited Bone Marrow Failure Syndromes (IBMFS) represent a group of devastating disorders where the genetic lesion directly impacts the bone marrow’s progenitor cells, resulting in pancytopenia (a deficiency of all three blood cell types). Fanconi Anemia (FA), for instance, is a highly heterogeneous disorder caused by mutations in any one of at least 22 different genes that form a complex pathway responsible for DNA repair. This underlying defect in genome maintenance leads to progressive bone marrow failure, high susceptibility to hematological malignancies like Acute Myeloid Leukemia (AML), and a variety of congenital abnormalities. Diamond-Blackfan Anemia (DBA) is another example, characterized by a pure red cell aplasia, meaning a severe lack of red blood cell precursors. Interestingly, DBA is often caused by mutations in ribosomal protein genes (e.g., ,
), implicating defects in ribosome biogenesis as a source of hematological failure. The connection here—between the cellular machinery for protein production and the survival of hematopoietic stem cells—is a striking illustration of the unexpected pathways through which genetic errors manifest as blood disease.
Fanconi Anemia (FA), for instance, is a highly heterogeneous disorder caused by mutations in any one of at least 22 different genes that form a complex pathway responsible for DNA repair.
The diagnostic landscape in hematological genetics has been utterly transformed by Next-Generation Sequencing (NGS) technologies. NGS, specifically panel sequencing and Whole-Exome Sequencing (WES), allows for the simultaneous analysis of hundreds of genes associated with various blood disorders, drastically reducing the time and cost associated with diagnosis. Prior to this, identifying a specific gene mutation in a heterogeneous disease like FA required laborious, sequential testing. Now, clinicians can rapidly pinpoint the causative variant, which is critical not only for confirming the diagnosis but also for informing personalized treatment plans. For instance, knowing the specific mutation in a myelodysplastic syndrome (MDS) patient might guide the choice between standard chemotherapy and a targeted therapy. This move towards high-throughput genetic profiling represents the current frontier in precision hematology, shifting the focus from treating symptoms to addressing the root molecular cause.
Now, clinicians can rapidly pinpoint the causative variant, which is critical not only for confirming the diagnosis but also for informing personalized treatment plans.
The profound understanding of the genetic basis of these disorders has naturally led to the development of radical, curative therapies. Gene therapy, a method of introducing a functional copy of a defective gene into a patient’s own cells, has seen remarkable breakthroughs. Clinical trials for both -thalassemia and Sickle Cell Disease are showing promising results where hematopoietic stem cells are harvested from the patient, genetically corrected ex vivo using viral vectors or CRISPR/Cas9 technology to replace or edit the faulty gene, and then re-infused. This process holds the potential to permanently correct the genetic defect, eliminating the need for chronic transfusions or the complexities of finding a matched donor for allogeneic stem cell transplantation. The initial successes, though not without their own technical and safety hurdles, herald a paradigm shift in how we conceptualize the treatment of monogenic blood disorders.
Gene therapy, a method of introducing a functional copy of a defective gene into a patient’s own cells, has seen remarkable breakthroughs.
Beyond inherited disorders, the genetics of acquired hematological malignancies—leukemias, lymphomas, and myelodysplastic syndromes—also relies heavily on understanding somatic mutations (those acquired during a lifetime). While not passed down through generations, these cancers arise from genetic mistakes in hematopoietic stem or progenitor cells. The identification of specific driver mutations, such as the –
fusion gene in Chronic Myeloid Leukemia (CML) or mutations in
and
in Acute Myeloid Leukemia (AML), has led to the development of highly effective targeted inhibitors. Imatinib, a tyrosine kinase inhibitor targeting BCR-ABL, dramatically changed CML from a fatal disease into a manageable chronic condition. This success story showcases the translational power of genetic research, where molecular identification immediately opens the door to rational drug design.
This success story showcases the translational power of genetic research, where molecular identification immediately opens the door to rational drug design.
The increasing availability of carrier screening and prenatal diagnostics presents a significant ethical and societal dimension to the genetics of blood disorders. Carrier screening allows prospective parents, particularly those from at-risk populations for conditions like SCD and thalassemia, to determine their risk of having an affected child. While offering reproductive choice and informing proactive measures, this raises complex questions about genetic privacy, stigmatization, and access to genetic counseling. The implementation of universal, population-based screening programs requires careful consideration of public health infrastructure, educational outreach, and the provision of non-directive counseling to ensure informed decision-making. The sheer volume of genetic data now available complicates every step, demanding a thoughtful, interdisciplinary approach that respects personal autonomy within the context of scientific capability.
The implementation of universal, population-based screening programs requires careful consideration of public health infrastructure, educational outreach, and the provision of non-directive counseling to ensure informed decision-making.
In final consideration, the field continues to discover new genes and novel pathogenic mechanisms. The emerging roles of non-coding RNAs and epigenetic modifications—changes that affect gene expression without altering the underlying DNA sequence—are adding layers of complexity to our understanding. For example, abnormal DNA methylation patterns are increasingly recognized as playing a role in both inherited conditions and acquired malignancies. The picture of blood disorders is therefore not a finished painting but a perpetually developing mural, where each new genetic discovery adds a crucial detail. The ongoing effort to connect a single-nucleotide polymorphism to a debilitating clinical outcome continues to drive the most promising avenues of therapeutic development, keeping personalized medicine at the very heart of modern hematology.